J. Chem. Phys. 135, art. no. 124712/8 pages (2011)
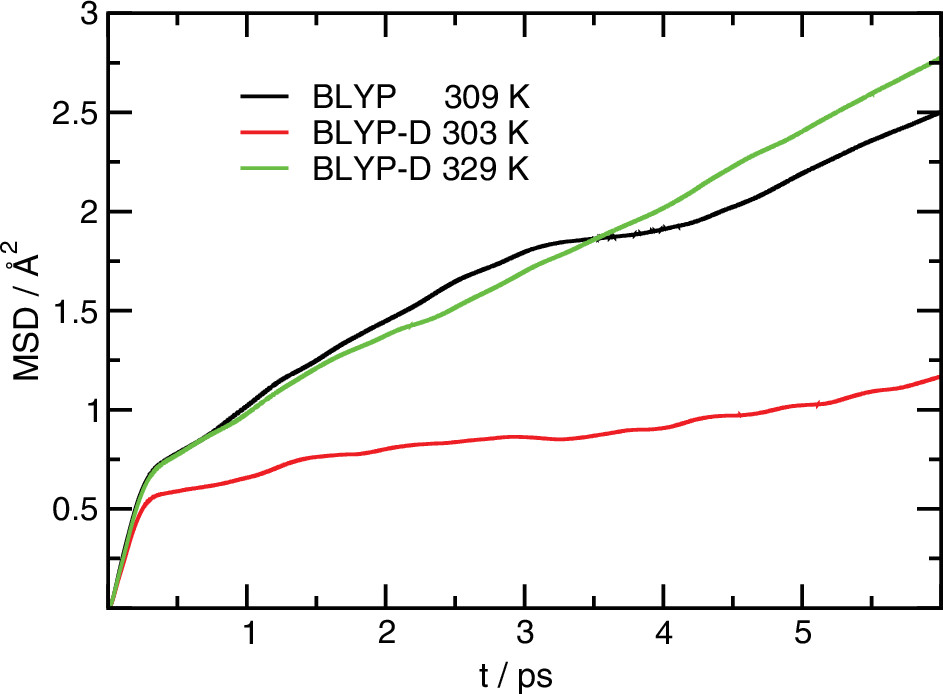
First-principles molecular dynamics simulations, in which the forces are computed from electronic structure calculations, have great potential to provide unique insight into structure, dynamics, electronic properties, and chemistry of interfacial systems that is not available from empirical force fields. The majority of current first-principles simulations are driven by forces derived from density functional theory with generalized gradient approximations to the exchange-correlation energy, which do not capture dispersion interactions. We have carried out first-principles molecular dynamics simulations of air-water interfaces employing a particular generalized gradient approximation to the exchange-correlation functional (BLYP), with and without empirical dispersion corrections. We assess the utility of the dispersion corrections by comparison of a variety of structural, dynamic, and thermodynamic properties of bulk and interfacial water with experimental data, as well as other first-principles and force field-based simulations.