Phys. Chem. Chem. Phys. 14, 4884-4890 (2012)
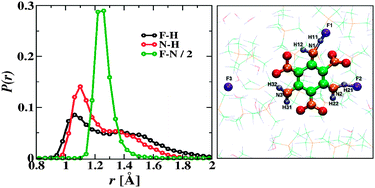
With the aim to understand the relatively high solubility of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), an important energetic material with a high degree of inter- and intra-molecular hydrogen bonding, in fluoride anion containing ionic liquids (ILs), first principles molecular dynamics simulations in the isobaric–isothermal ensemble were carried out for a system using hydrous tetramethylammonium fluoride as the prototypical solvent. Simulations initiated from both molecular TATB and its Meisenheimer complex (i.e., a σ-complex of the fluoride and the electrophilic ring of TATB) yield a Zundel-type complex where a proton is shared between an amino group and an F− ion, whereas the Meisenheimer complex is found to be only transiently stable. An analysis of the electronic structure probing the Wannier function centers supports the finding of a proton-sharing complex with a three-center four-electron like bond. The Zundel-type complex also yields an electronic absorption spectrum consistent with the experimentally observed color change. This study provides evidence that the remarkable solubility of otherwise hard-to-dissolve molecular crystals in ILs can be aided by chemical modification of the solute.