Theor. Chem. Acc. 132, art. no. 1388/16 pages (2013)
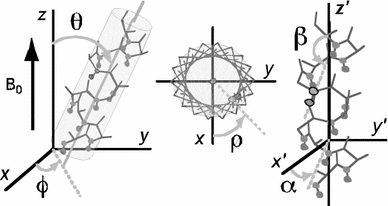
We present a fast and simple protocol to obtain moderate-resolution backbone structures of helical proteins. This approach utilizes a combination of sparse backbone NMR data (residual dipolar couplings and paramagnetic relaxation enhancements) or EPR data with a residue-based force field and Monte Carlo/simulated annealing protocol to explore the folding energy landscape of helical proteins. By using only backbone NMR data, which are relatively easy to collect and analyze, and strategically placed spin relaxation probes, we show that it is possible to obtain protein structures with correct helical topology and backbone RMS deviations well below 4 Å. This approach offers promising alternatives for the structural determination of proteins in which nuclear Overhauser effect data are difficult or impossible to assign and produces initial models that will speed up the high-resolution structure determination by NMR spectroscopy.