J. Phys. Chem. C 120, 18707-18712 (2016)
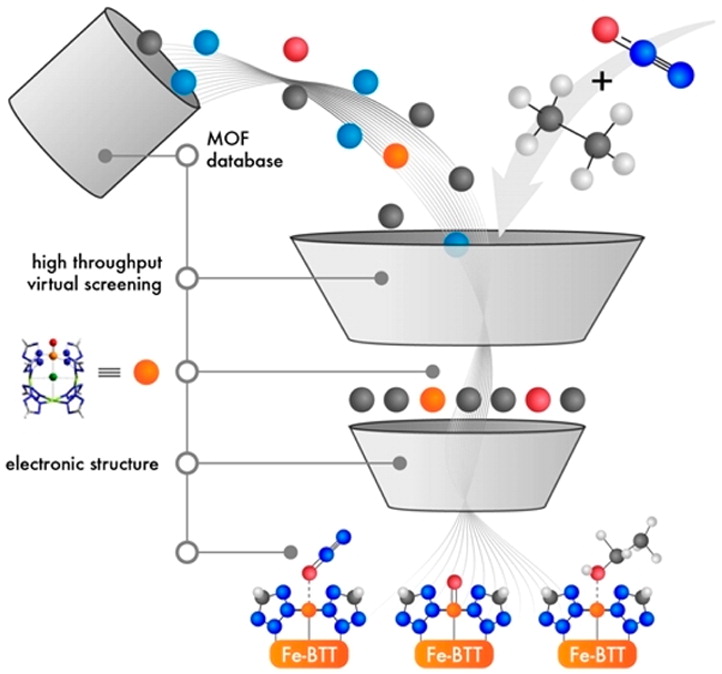
High-spin iron(IV)–oxo compounds are known to activate strong C–H bonds. Stabilizing the high-spin S = 2 electronic configuration is difficult in molecular species for homogeneous catalysis, but recent experimental and computational results suggest this can be achieved in the metal–organic framework Fe2(dobdc) (dobdc4– = 2,5-dioxido-1,4-benzenedicarboxylate) and its magnesium-diluted analogues. With a novel computational screening approach, we have identified three additional frameworks that are predicted to form high-spin iron(IV)–oxo species upon dissociative adsorption of nitrous oxide. The computational work is supported by follow-up experiments which show that, among these three materials, Fe-BTT (BTT3– = 1,3,5-benzenetristetrazolate) selectively oxidizes ethane to ethanol at 120 °C. Subsequent spectroscopic and cycling studies suggest that framework defects, rather than the bulk framework or extraframework sites, are likely responsible for the observed reactivity. This work shows how computational methods can be used to rapidly identify promising candidate frameworks, and highlights the need for new methods that allow defect sites in metal–organic frameworks to be better understood and exploited for catalysis.