J. Chem. Phys. 102, 2126-2140 (1995); 'Erratum', 109, 352 (1998)
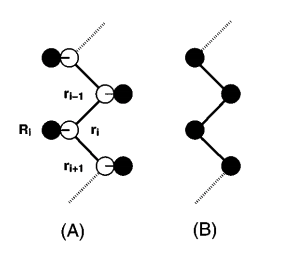
For petrochemical applications knowledge of the critical properties of the n‐alkanes is of interest even at temperatures where these molecules are thermally unstable. Computer simulations can determine the vapor–liquid coexistence curve of a large number of n‐alkanes ranging from pentane (C5) through octatetracontane (C48). We have compared the predicted phase diagrams of various models with experimental data. Models which give nearly identical properties of liquid alkanes at standard conditions may have critical temperatures that differ by more than 100 K. A new n‐alkane model has been developed by us that gives a good description of the phase behavior over a large temperature range. For modeling vapor–liquid coexistence a relatively simple united atom model was sufficient to obtain a very good agreement with experimental data; thus it appears not necessary to take the hydrogen atoms explicitly into account. The model developed in this work has been used to determine the critical properties of the long‐chain alkanes for which experiments turned out to be difficult and contradictory. We found that for the long‐chain alkanes (C8–C48) the critical density decreases as a function of the carbon number. These simulations were made possible by the use of a recently developed simulation technique, which is a combination of the Gibbs‐ensemble technique and the configurational‐bias Monte Carlo method. Compared with the conventional Gibbs‐ensemble technique, this method is several orders of magnitude more efficient for pentane and up to a hundred orders of magnitude for octatetracontane. This recent development makes it possible to perform routinely phase equilibrium calculations of complex molecules.