J. Phys. Chem. C 2021, 125, 11, 6090–6098
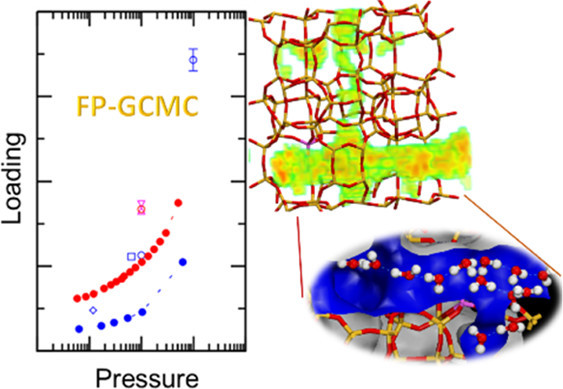
Water appears by design or as impurities in many important reactive systems. For those catalyzed by porous solid acids, such as widely used zeolites, experimentally quantifying the amount or elucidating the structure of adsorbed water clusters at reaction conditions is challenging, while computational studies (e.g., first-principles molecular dynamics simulations) often need to assume the loading to examine solvation effects. Here, we perform first-principles grand-canonical simulations to predict water adsorption to H-ZSM-5 zeolites under specified experimental conditions. Presampling with inexpensive force fields and a pool-based parallelization algorithm are used to improve simulation efficiency, while molecular dynamics is used to sample configurations involving hydronium species. We observe that H+ exchange dramatically increases the hydrophilicity of zeolite MFI and an appreciable amount of water is present at very low relative humidities. At all conditions examined, the zeolitic protons are found to dissociate readily from the surface basic sites and become mobile by participating in the hydrogen-bonded chains of adsorbed water molecules.