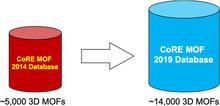
Publications 2010 - 2019
[228] Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019
J. Chem. Eng. Data 64, 5985–5998 (2019)
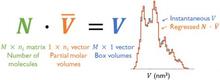
[227] Partial molar properties from molecular simulation using multiple linear regression
Mol. Phys. 117, 3589–3602 (2019)
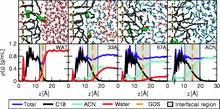
[226] Computational screening of metal–organic frameworks for biogas purification
Mol. Syst. Des. Eng. 4, 1125–1135 (2019)
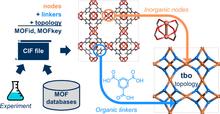
[225] Identification Schemes for Metal–Organic Frameworks To Enable Rapid Search and Cheminformatics Analysis
Cryst. Growth Des. 19, 6682–6697 (2019)
[224] Molecular Simulations Probing the Thermophysical Properties of Homogeneously Stretched and Bubbly Water Systems
J. Chem. Eng. Data 64, 3755–3771 (2019)

[223] Synthesis, Simulation, and Self-Assembly of a Model Amphiphile To Push the Limits of Block Polymer Nanopatterning
Nano Lett. 19, 4458–4462 (2019)
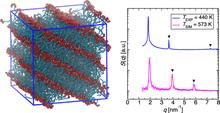
[222] Metal–Organic Frameworks with Metal–Catecholates for O2/N2 Separation
J. Phys. Chem. C 123, 12935–12946 (2019)
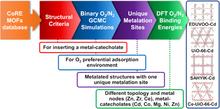
[221] Deep neural network learning of complex binary sorption equilibria from molecular simulation data
Chem. Sci. 10, 4377–4388 (2019)
[220] A new equation of state for homo-polymers in dissipative particle dynamics
J. Chem. Phys. 150, 124104/13 pages (2019)
[219] Column selection for comprehensive two-dimensional liquid chromatography using the hydrophobic subtraction model
J. Chromtogr. A 1589, 47–55 (2019)

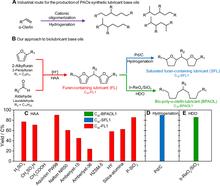
[218] Renewable lubricants with tailored molecular architecture
Sci. Adv. 5, eaav5487 (2019)
[217] Monte Carlo Simulations of Fluid Phase Equilibria and Interfacial Properties for Water/Alkane Mixtures: An Assessment of Nonpolarizable Water Models and of Departures from the Lorentz–Berthelot Combining Rules
J. Chem. Eng. Data 63, 4256–4268 (2018)
[216] Using molecular simulations to probe pore structures and polymer partitioning in size exclusion chromatography
J. Chromtogr. A 1573, 78–86 (2018)
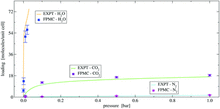
[215] First principles Monte Carlo simulations of unary and binary adsorption: CO2, N2, and H2O in Mg-MOF-74
Chem. Comm. 54, 10816–10819 (2018)
[214] A Monte Carlo simulation study of the interfacial tension for water/oil mixtures at elevated temperatures and pressures: Water/n-dodecane, water/toluene, and water/(n-dodecane + toluene)
Fluid Phase Equil. 476, 16–24 (2018)

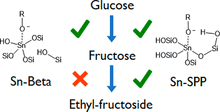
[213] Cooperative Catalysis by Surface Lewis Acid/Silanol for Selective Fructose Etherification on Sn-SPP Zeolite
ACS Catal. 8, 9056–9065 (2018)
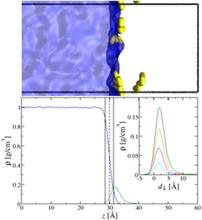
[212] Monte Carlo simulations probing the liquid/vapour interface of water/hexane mixtures: adsorption thermodynamics, hydrophobic effect, and structural analysis
Mol. Phys. 116, 3283–3291 (2018)
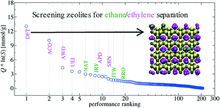
[211] C2 adsorption in zeolites: in silico screening and sensitivity to molecular models
Mol. Syst. Des. Eng. 3, 619–626 (2018)
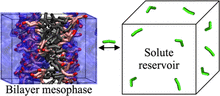
[210] Probing Additive Loading in the Lamellar Phase of a Nonionic Surfactant: Gibbs Ensemble Monte Carlo Simulations Using the SDK Force Field
Langmuir 34, 8245–8254 (2018)
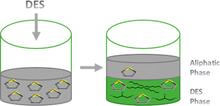
[209] Mercury Capture from Petroleum Using Deep Eutectic Solvents
Ind. Eng. Chem. Res. 57, 9222–9230 (2018)

[208] Understanding the unique sorption of alkane-α, ω-diols in silicalite-1
J. Chem. Phys. 149, art. no. 072331/17 pages (2018)
[207] Understanding the Molecular Weight Dependence of χ and the Effect of Dispersity on Polymer Blend Phase Diagrams
Macromolecules 51, 3774-3787 (2018)

[206] Computational Design of High-χ Block Oligomers for Accessing 1 nm Domains
ACS Nano 12, 4351-4361 (2018)

[205] Supersaturated calcium carbonate solutions are classical
Sci. Adv. 4, art. no. eaao6283/11 pages (2018)
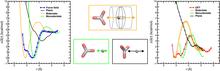
[204] Efficient calculation of absorption spectra in solution: Approaches for selecting representative solvent configurations and for reducing the number of explicit solvent molecules
Acta Phys. Chem. Sin. 34, 1106-1115 (2018)
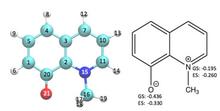
[203] Understanding the Reactive Adsorption of H2S and CO2 in Sodium‐Exchanged Zeolites
ChemPhysChem 19, 512-518 (2018)

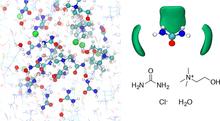
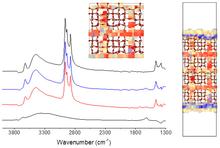
[202] First-Principles Molecular Dynamics Study of a Deep Eutectic Solvent: Choline Chloride/Urea and Its Mixture with Water
J. Phys. Chem. B 122, 1245-1254 (2018)
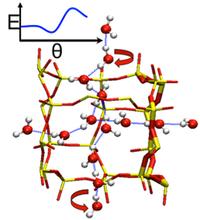
[201] Deconstructing the Confinement Effect upon the Organization and Dynamics of Water in Hydrophobic Nanoporous Materials: Lessons Learned from Zeolites
J. Phys. Chem. C 121, 22015-22024 (2017)
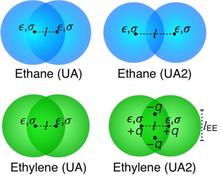
[200] Transferable potentials for phase equilibria. Improved united‐atom description of ethane and ethylene
AIChE J. 63, 5098-5110 (2017)
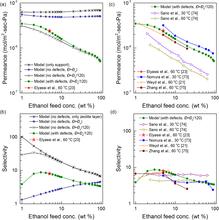
[199] Bioethanol enrichment using zeolite membranes: Molecular modeling, conceptual process design and techno-economic analysis
J. Membrane Sci. 540, 464-476 (2017)
[198] Separation of Thiophene from Aliphatic Hydrocarbons Using Tetrahexylammonium-Based Deep Eutectic Solvents as Extracting Agents
J. Chem. Eng. Data 62, 2911-2919
[197] Comparative Study of the Effect of Defects on Selective Adsorption of Butanol from Butanol/Water Binary Vapor Mixtures in Silicalite-1 Films
Langmuir 33, 8420-8427 (2017)
[196] Computational Screening of Nanoporous Materials for Hexane and Heptane Isomer Separation
Chem. Mater. 29, 6315-6328 (2017)
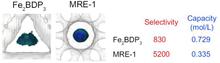
[195] Sub-Micrometer Zeolite Films on Gold-Coated Silicon Wafers with Single-Crystal-Like Dielectric Constant and Elastic Modulus
Adv. Funct. Mater. 27, 1700864 (2017)
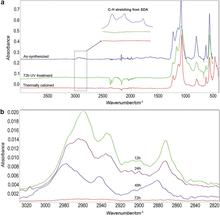
[194] Ultra-selective high-flux membranes from directly synthesized zeolite nanosheets
Nature 543, 690-694 (2017)
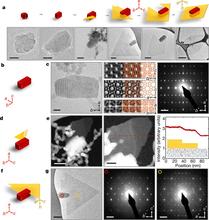
[193] Using the k-d Tree Data Structure to Accelerate Monte Carlo Simulations
J. Chem. Theor. Comp. 13, 1556-1565 (2017)

[192] Assessment and Optimization of Configurational-Bias Monte Carlo Particle Swap Strategies for Simulations of Water in the Gibbs Ensemble
J. Chem. Theor. Comp. 13, 431-440 (2017)

[191] A mathematical model for zeolite membrane module performance and its use for techno-economic evaluation of improved energy efficiency hybrid membrane-distillation processes for butane isomer separations
J. Membr. Sci. 520, 434-449 (2016)
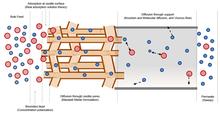
[190] Accelerated Computational Analysis of Metal–Organic Frameworks for Oxidation Catalysis
J. Phys. Chem. C 120, 18707-18712 (2016)
[189] Understanding Diffusion in Hierarchical Zeolites with House-of-Cards Nanosheets
ACS Nano 10, 7612-7618 (2016)

[188] First-Principles Monte Carlo Simulations of Reaction Equilibria in Compressed Vapors
ACS Central Science 2, 409-415 (2016)
[187] CO2 Adsorption in M-IRMOF-10 (M = Mg, Ca, Fe, Cu, Zn, Ge, Sr, Cd, Sn, Ba)
J. Phys. Chem. C 120, 12819-12830 (2016)
[186] Molecular Simulation of Olefin Oligomer Blend Phase Behavior
Macromolecules 49, 3975-3985 (2016)
[185] Identifying Optimal Zeolitic Sorbents for Sweetening of Highly Sour Natural Gas
Angew. Chem. Intl. Ed. 55, 5938-5942 (2016)
[184] Structure and Phase Behavior of Mixed Self-Assembled Alkanethiolate Monolayers on Gold Nanoparticles: A Monte Carlo Study
J. Phys. Chem. B 120, 1972-1978 (2016)
[183] Adsorptive Separation of 1-Butanol from Aqueous Solutions Using MFI- and FER-Type Zeolite Frameworks: A Monte Carlo Study
Langmuir 32, 2093-2101 (2016)
[182] A Monte Carlo simulation study of the liquid–liquid equilibria for binary dodecane/ethanol and ternary dodecane/ethanol/water mixtures
Fluid Phase Equil. 407, 269-279 (2016)
[181] Monte Carlo Simulations Probing the Adsorptive Separation of Hydrogen Sulfide/Methane Mixtures Using All-Silica Zeolites
Langmuir 31, 12268-12278 (2015)

[180] Understanding the sensitivity of nucleation free energies: The role of supersaturation and temperature
J. Chem. Phys. 143, art. no. 164516/6 pages (2015)
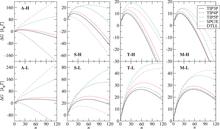
[179] Accurate and precise determination of critical properties from Gibbs ensemble Monte Carlo simulations
J. Chem. Phys. 143, art. no. 114113/13 pages (2015)
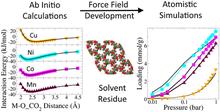
[178] Ab Initio Derived Force Fields for Predicting CO2 Adsorption and Accessibility of Metal Sites in the Metal–Organic Frameworks M-MOF-74 (M = Mn, Co, Ni, Cu)
J. Phys. Chem. C 119, 16058-16071 (2015)
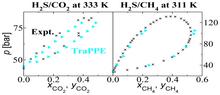
[177] Development of the Transferable Potentials for Phase Equilibria Model for Hydrogen Sulfide
J. Phys. Chem. B 119, 7041-7052 (2015)
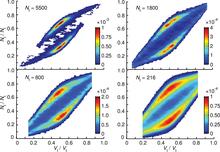
[176] Discovery of optimal zeolites for challenging separations and chemical transformations using predictive materials modeling
Nat. Commun. 6, art. no. 5912/9 pages (2015)
[175] Liquid–liquid equilibria for soft-repulsive particles: Improved equation of state and methodology for representing molecules of different sizes and chemistry in dissipative particle dynamics
J. Chem. Phys. 142, art. no. 044902/13 pages (2015)
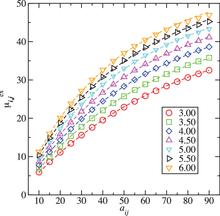
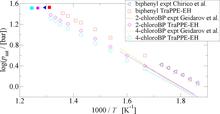
[174] Prediction of Vapor–Liquid Coexistence Properties and Critical Points of Polychlorinated Biphenyls from Monte Carlo Simulations with the TraPPE–EH Force Field
J. Chem. Eng. Data 59, 3301-3306 (2014)
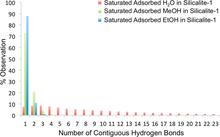
[173] Deconstructing Hydrogen-Bond Networks in Confined Nanoporous Materials: Implications for Alcohol–Water Separation
J. Phys. Chem. C 118, 19723-19732 (2014)
[172] Understanding the Unusual Adsorption Behavior in Hierarchical Zeolite Nanosheets
ChemPhysChem 15, 2225-2229 (2014)

[171] Monte Carlo Simulations of Thin Hydrocarbon Films: Composition Heterogeneity and Structure at the Solid–Liquid and Liquid–Vapor Interfaces
Langmuir 30, 3086-3094 (2014)

[170] An online parameter and property database for the TraPPE force field
Molec. Simul. 40, 101-105 (2014)

[169] A computational study of the adsorption of n-perfluorohexane in zeolite BCR-704
Fluid Phase Equil. 366, 146-151 (2014)
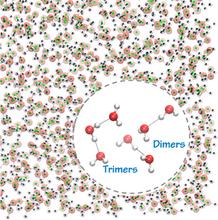
[168] Water 26-mers Drawn from Bulk Simulations: Benchmark Binding Energies for Unprecedentedly Large Water Clusters and Assessment of the Electrostatically Embedded Three-Body and Pairwise Additive Approximations
J. Phys. Chem. Lett. 5, 666-670 (2014)
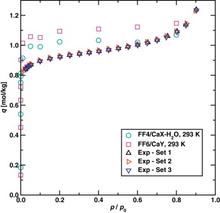
[167] Concentration effects on the selective extraction of ethanol from aqueous solution using silicalite-1 and decanol isomers
Fluid Phase Equil. 362, 118-124 (2014)
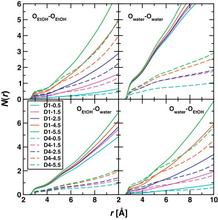
[166] TraPPE-zeo: Transferable Potentials for Phase Equilibria Force Field for All-Silica Zeolites
J. Phys. Chem. C 117, 24375-24387 (2013)
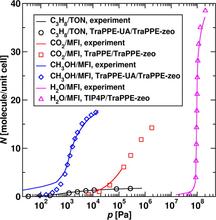
[165] Influence of simulation protocols on the efficiency of Gibbs ensemble Monte Carlo simulations
Molec. Simul. 39, 1135-1142 (2013)
[164] Modeling helical proteins using residual dipolar couplings, sparse long-range distance constraints and a simple residue-based force field
Theor. Chem. Acc. 132, art. no. 1388/16 pages (2013)
[163] Adsorption of glucose into zeolite beta from aqueous solution
AIChE J. 59, 3523-3529 (2013)
[162] Calculation of the Gibbs free energy of solvation and dissociation of HCl in water via Monte Carlo simulations and continuum solvation models
Phys. Chem. Chem. Phys. 15, 13578-13585 (2013)
[161] Molecular insights for the optimization of solvent‐based selective extraction of ethanol from fermentation broths
AIChE J. 59, 3065-3070 (2013)
[160] Selective adsorption from dilute solutions: Gibbs ensemble Monte Carlo simulations
Fluid Phase Equil. 351, 1-6 (2013)
[159] Energetics of Atmospherically Implicated Clusters Made of Sulfuric Acid, Ammonia, and Dimethyl Amine
J. Phys. Chem. A 117, 3819-3825 (2013)
[158] Transferable Potentials for Phase Equilibria. 10. Explicit-Hydrogen Description of Substituted Benzenes and Polycyclic Aromatic Compounds
J. Phys. Chem. B 117, 273-288 (2013)
[157] Efficient methods for including quantum effects in Monte Carlo calculations of large systems: Extension of the displaced points path integral method and other effective potential methods to calculate properties and distributions
J. Chem. Phys. 138, art. no. 014110/15 pages (2013)
[156] Acid–base chemical reaction model for nucleation rates in the polluted atmospheric boundary layer
Proc. Natl. Acad. Sci. USA 109, 18713-18718 (2012)
[155] Multicomponent Adsorption of Alcohols onto Silicalite-1 from Aqueous Solution: Isotherms, Structural Analysis, and Assessment of Ideal Adsorbed Solution Theory
Langmuir 28, 15566-15576 (2012)
[154] Transferable Potentials for Phase Equilibria–United Atom Description of Five- and Six-Membered Cyclic Alkanes and Ethers
J. Phys. Chem. B 116, 11234-11246 (2012)
[153] Electrostatically embedded many-body method for dipole moments, partial atomic charges, and charge transfer
Phys. Chem. Chem. Phys. 14, 7669-7678 (2012)
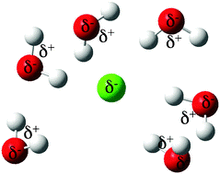
[152] Understanding the solubility of triamino-trinitrobenzene in hydrous tetramethylammonium fluoride: a first principles molecular dynamics simulation study
Phys. Chem. Chem. Phys. 14, 4884-4890 (2012)
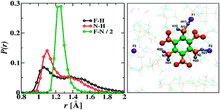
[151] A molecular simulation study of the effects of stationary phase and solute chain length in reversed-phase liquid chromatography
J. Chromatogr. A 1223, 24-34 (2012)
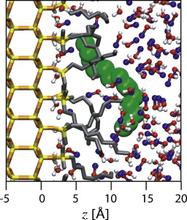
[150] Retention mechanism for polycyclic aromatic hydrocarbons in reversed-phase liquid chromatography with monomeric stationary phases
J. Chromatogr. A 1218, 9183-9193 (2011)
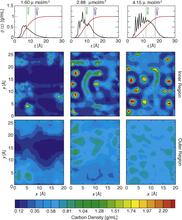
[149] Liquid structures of water, methanol, and hydrogen fluoride at ambient conditions from first principles molecular dynamics simulations with a dispersion corrected density functional
Phys. Chem. Chem. Phys. 13, 19943-19950 (2011)
[148] Vapor–Liquid Coexistence Curves for Methanol and Methane Using Dispersion-Corrected Density Functional Theory
J. Phys. Chem. B 115, 11688-11692 (2011)
[147] Re-examining the properties of the aqueous vapor–liquid interface using dispersion corrected density functional theory
J. Chem. Phys. 135, art. no. 124712/8 pages (2011)
[146] Phase Diagram of Water under an Applied Electric Field
Phys. Rev. Lett. 107, art. no. 155702/4 pages (2011)
[145] Correlations for Adsorption of Oxygenates onto Zeolites from Aqueous Solutions
J. Phys. Chem. B 115, 11431-11438 (2011)
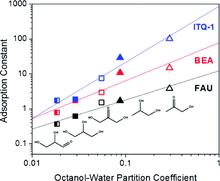
[144] Gibbs ensemble Monte Carlo simulations for the liquid–liquid phase equilibria of dipropylene glycol dimethyl ether and water: A preliminary report
Fluid Phase Equil. 310, 11-18 (2011)

[143] Assessing group-based cutoffs and the Ewald method for electrostatic interactions in clusters and in saturated, superheated, and supersaturated vapor phases of dipolar molecules
Theor. Chem. Acc. 130, 83-93 (2011)
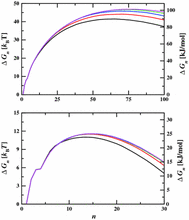
[R27] Hydrogen Sulfide Capture: From Absorption in Polar Liquids to Oxide, Zeolite, and Metal–Organic Framework Adsorbents and Membranes
Chem. Rev. 117, 9755-9803 (2017)
[R26] A Review of Biorefinery Separations for Bioproduct Production via Thermocatalytic Processing
Annu. Rev. Chem. Biomol. 8, 115-137 (2017)
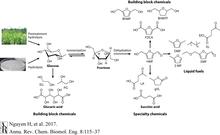
[R25] How Reversed-Phase Liquid Chromatography Works
LC GC North America 31, 630-637 (2013)
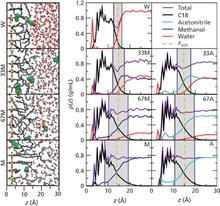
[R24] Molecular simulation studies of reversed-phase liquid chromatography
J. Chromatogr. A 1287, 60-82 (2013)
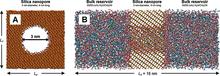
[R23] Molecular Simulations of Retention in Chromatographic Systems: Use of Biased Monte Carlo Techniques to Access Multiple Time and Length Scales
Topics in Current Chemistry, Vol. 307 Eds. B. Kirchner and J. Vrabec, Springer: Berlin, 2012, pp. 181-200