J. Chromatogr. A 1287, 60-82 (2013)
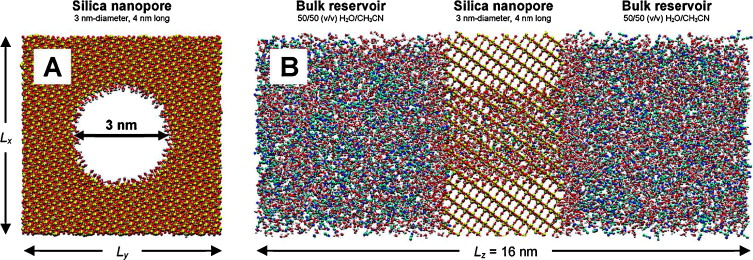
Over the past 20 years, molecular simulation methods have been applied to the modeling of reversed-phase liquid chromatography (RPLC). The purpose of these simulations was to provide a molecular-level understanding of: (i) the structure and dynamics of the bonded phase and its interface with the mobile phase, (ii) the interactions of analytes with the bonded phase, and (iii) the retention mechanism for different analytes. However, the investigation of chromatographic systems poses significant challenges for simulations with respect to the accuracy of the molecular mechanics force fields and the efficiency of the sampling algorithms. This review discusses a number of aspects concerning molecular simulation studies of RPLC systems including the historical development of the subject, the background needed to understand the two prevalent techniques, molecular dynamics (MD) and Monte Carlo (MC) methods, and the wealth of insight provided by these simulations. Examples from the literature employing MD approaches and from the authors’ laboratory using MC methods are discussed. The former can provide information on chain dynamics and transport properties, whereas the latter techniques are uniquely suited for the investigation of phase and sorption equilibria that underly RPLC retention, and both can be used to elucidate the bonded-chain conformations and solvent distributions.