Macromolecules 49, 3975-3985 (2016)
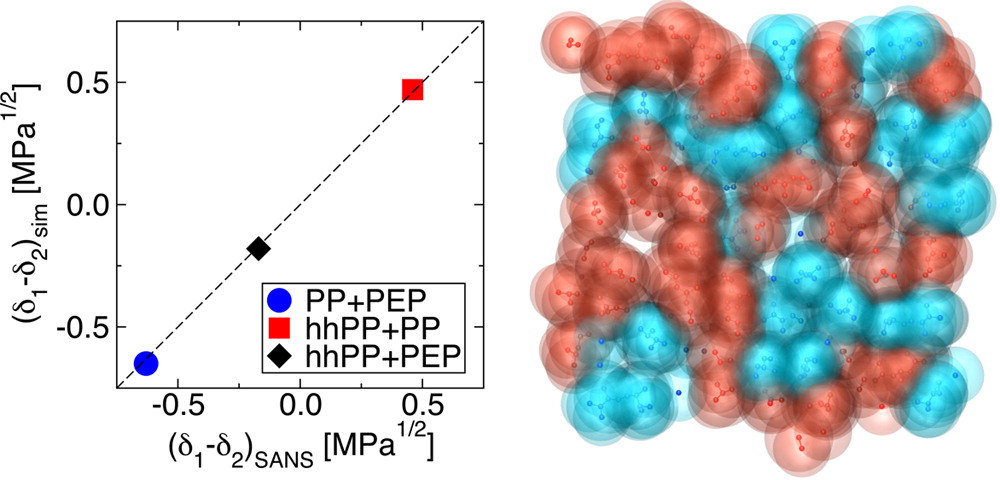
Configurational-bias Monte Carlo simulations in the Gibbs ensemble are used to study the thermodynamic and structural properties associated with the miscibility of binary olefin oligomer mixtures representing poly(ethylene-alt-propylene), polypropylene, and head-to-head polypropylene. Single-component simulations are performed to compute the cohesive energy densities, ΠCED, of different oligomers that are often utilized in estimating the miscibilities of compounds in the liquid phase but are not measurable for high-boiling compounds, such as polymers. Extrapolating simulation data for C5 to C36 oligomers allows for determination of the infinite-chain-length ΠCED values of three polyolefins. The results agree remarkably well with values deduced from small-angle neutron scattering experiments on high-molecular-weight polymers. In addition, the Flory–Huggins χ parameters based on the free energy of mixing for pairs of olefins are calculated directly from simulations of binary mixtures. The binary propylene and head-to-head propylene oligomer blend is found to exhibit stabilized irregular mixing behavior, in agreement with its polymeric counterpart. This chain-length independence of the mixing behavior is interpreted via insights from structural analysis. Our results identify simulations of oligomeric systems as a promising route to predict and understand polymer blend phase behavior.