J. Phys. Chem. C 120, 12819-12830 (2016)
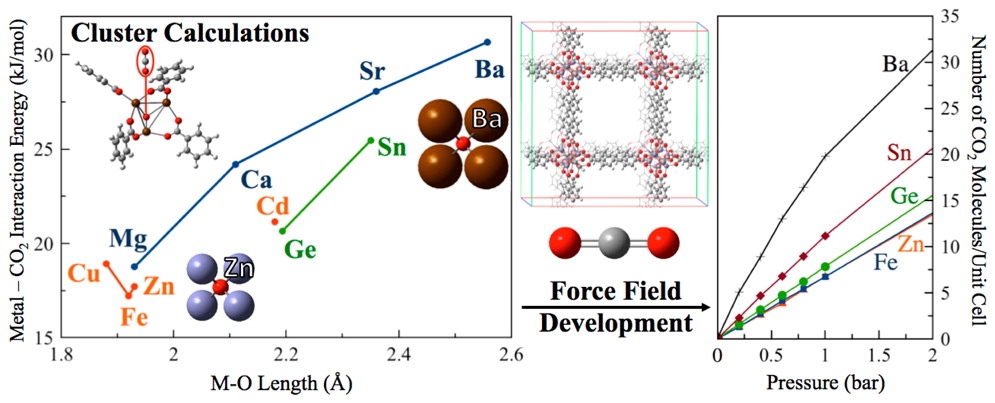
Metal–organic frameworks (MOFs) have been studied extensively for application in flue gas separation because of their tunability, structural stability, and large surface area. M-IRMOF-10 (M = transition metal or main-group atom) is a well-studied series of structures and is composed of saturated tetrahedral Zn4O nodes and dicarboxylate linkers that form a cubic unit cell. We report the results of a computational study on the effects that changing the metal atoms within IRMOF-10 has on the affinity of the material towards CO2. Force fields were parametrized using quantum mechanical calculations to systematically compare the effects of different metal centers on CO2 adsorption at high and low pressure. Two different methods for the determination of partial charges (DDEC and CM5) and force field parameter sets (TraPPE and UFF) were employed. TraPPE parameters with fitted metal–CO2 interactions and CM5 charges resulted in isotherms that were closer to experiment than pure UFF. The results indicate that exchanging the Zn2+ ions in the IRMOF-10 series with metals that have larger ionic radii (Sn2+ and Ba2+) can lead to an increase in CO2 affinity due to the increased exposure of the positive metal charge to the oxygen atoms of CO2 and the increased interaction from the more diffuse electrons.