Theor. Chem. Acc. 130, 83-93 (2011)
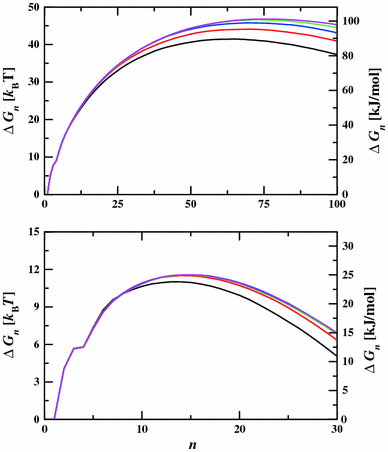
Monte Carlo simulations in the canonical, isobaric-isothermal, grand canonical, and Gibbs ensembles were used to assess whether the computationally expensive Ewald summation method for the computation of the first-order electrostatic energy can be replaced with a simpler truncation approach for accurate simulations of the saturated, superheated, and supersaturated vapor phases of dipolar and hydrogen-bonding molecules. Rotationally averaged hydrogen fluoride dimer and trimer energies, thermophysical properties and aggregation in the superheated vapor phase of hydrogen fluoride, nucleation free energy barriers for water, and the vapor–liquid coexistence properties of hydrogen fluoride and water were investigated over a wide range of state points. We find that for densities not too close to the critical density, results obtained from simulations using a spherical potential truncation based on neutral groups (molecules or fragments) for the Coulomb interactions are statistically identical to those obtained using the Ewald summation method. Use of the simpler spherical truncation results in a significant reduction of the computational effort for simulations employing molecular mechanics force fields and also allows for straightforward implementation of many-body expansion methods to compute the potential energy from electronic structure calculations of subsystems of the entire vapor-phase system.