[107] Prediction of viscosities and vapor-liquid equilibria for five polyhydric alcohols by molecular simulation
Fluid Phase Equil. 260, 218-231 (2007)
Publication Abstract
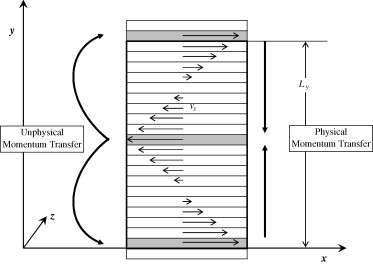
Reverse nonequilibrium molecular dynamics in the canonical ensemble and coupled–decoupled configurational-bias Monte Carlo simulations in the Gibbs ensemble were used to predict the low-shear rate Newtonian viscosities and vapor–liquid coexistence curves for 1,2-butanediol, 1,3-butanediol, 1,4-butanediol, 2-methyl-1,3-propanediol, and 1,2,4-butanetriol modeled with the transferable potentials for phase equilibria-united atom (TraPPE-UA) force field. Comparison with available experimental data demonstrates that the TraPPE-UA force field yields very good predictions of the viscosities and vapor–liquid coexistence curves. A detailed analysis of liquid structure and hydrogen bonding is provided.