J. Phys. Chem. C 111, 16227-16242 (2007)
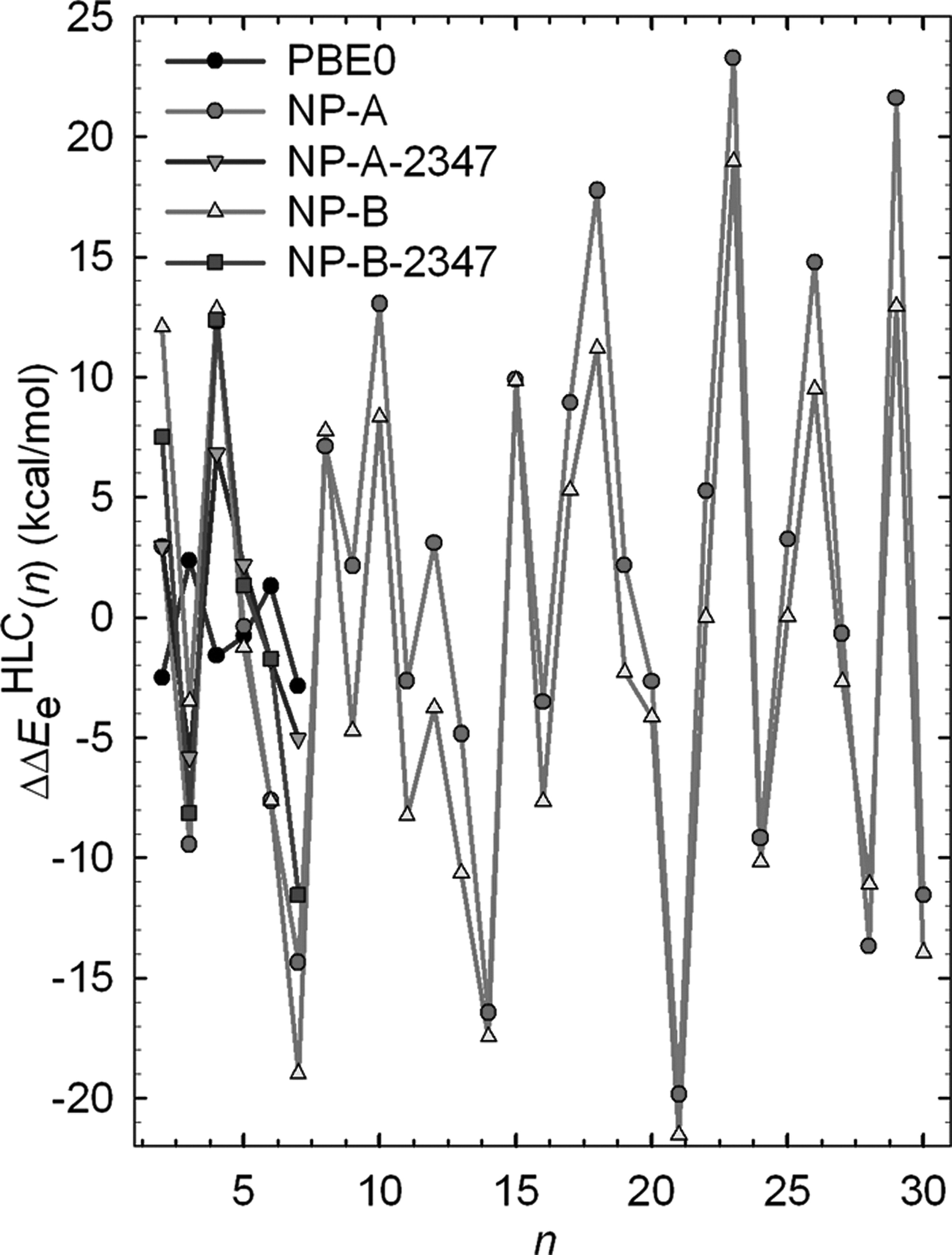
Efficient simulation methods are presented for determining the standard Gibbs free energy changes for the reactions, M + Mn-1 ↔ Mn (R1), involved in the formation of atomic clusters and nanoparticles (also called particles) in the vapor phase. The standard Gibbs free energy of formation (ΔfG°) of a particle is obtained from these Gibbs free energy changes (ΔG°) by a recursion relationship using the experimental ΔfG° of the monomer. In the present study, this method has been applied to reactions involving Aln particles with n = 2−60. This method has been validated for n = 2, where the experimental thermodynamic properties of Al2 have been recompiled using the latest available experimental or highly accurate theoretical data. For n = 2−4, two completely different approaches, a Monte Carlo configuration integral (MCCI) integration of partition functions and a Monte Carlo direct simulation of the equilibrium constants (MCEC), employing four well-validated potential energy functions have been used to calculate ΔG° of R1. Excellent agreement is observed for these two methods. Although different potential energy functions give different stage-1 results for n ≤ 10, three high-level correction (HLC) terms, namely, a correction for the potential energy difference of the global minima, another for the electronic excitation contribution, and a third based on calculating isomeric−rovibrational contribution, have been applied to mitigate deficiencies in the potential energy functions. For n = 2, good agreement has been found between the corrected simulation results and experimental data. For larger n, the more efficient MCEC method has been used. Finally, accurate ΔG° of R1 and thus ΔfG° of Aln particles with n = 2−60 have been determined. This is the first example of the determination of nanoparticle free energies of formation.