J. Chem. Phys. 129, art. no. 194510/8 pages (2008)
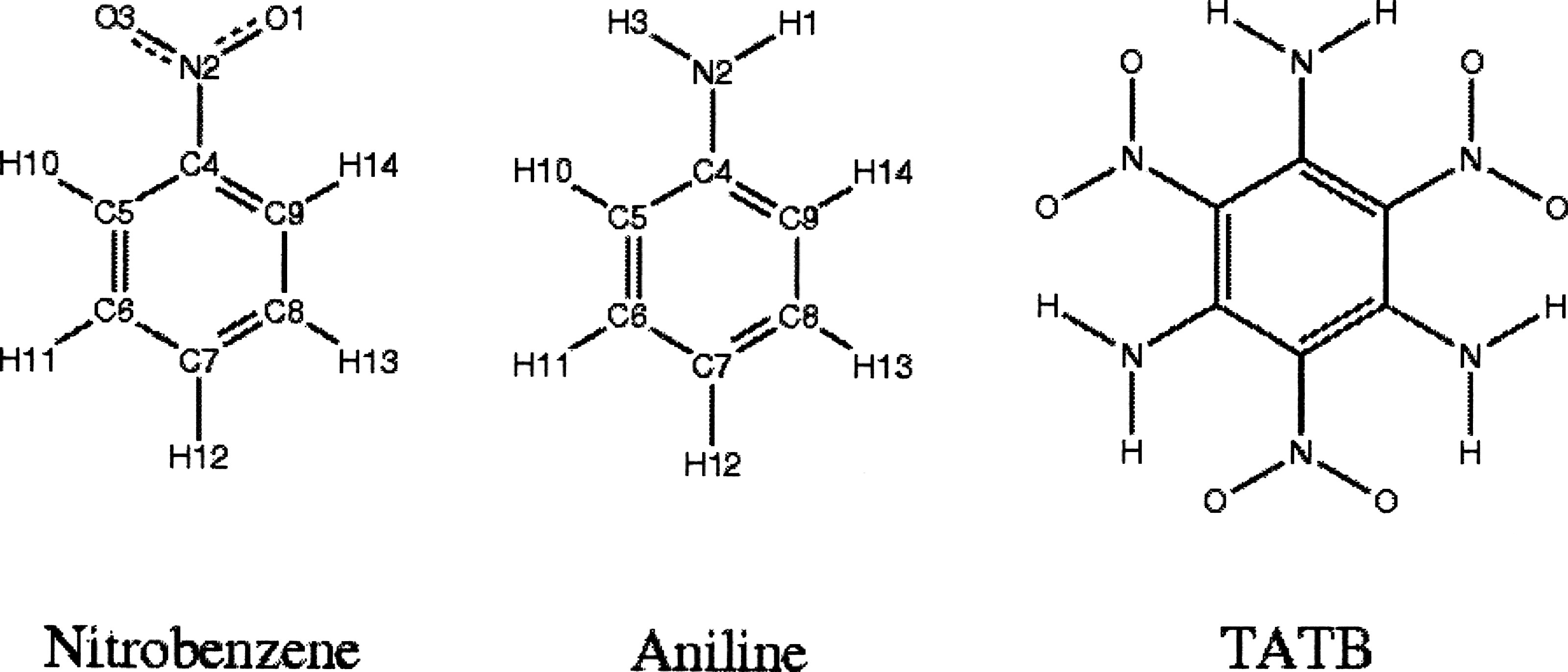
The transferable potentials for phase equilibria (TraPPE) force field was extended to nitro and amino substituents for aromatic rings via parametrization to the vapor-liquid coexistence curves of nitrobenzene and aniline, respectively. These groups were then transferred to model 1,3,5-triamino-2,4,6-trinitrobenzene (TATB). Without any further parametrization to solid state data, the TraPPE force field is able to predict TATB’s unit cell lengths and angles at 295K295K with mean unsigned percentage errors of 0.3% and 1.8% and the specific density within 0.5%. These predictions are comparable in accuracy to the GRBF model [Gee et al., J. Chem. Phys. 120, 7059 (2004)] that was parametrized directly to TATB’s solid state properties. Both force fields are able to reproduce the pressure dependence of TATB’s unit cell volume, but they underestimate its thermal expansion. Due to its energetic nature and unusually large cohesive energy, TATB is not chemically stable at temperature in its liquid range. Gibbs ensemble simulations allow one to determine TATB’s vapor-liquid coexistence curve at elevated temperatures and the predicted critical temperature and density for the TraPPE and GRBF model are 937±8 and 1034±8K, and 0.52±0.02 and 0.50±0.02g∕cm3, respectively.