J. Phys. Chem. C 113, 10354-10370 (2009)
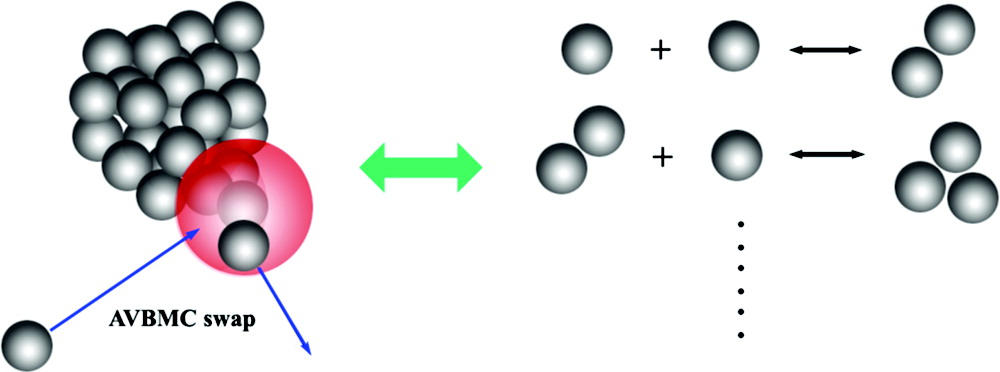
Nucleation of new particles from vapor-phase molecular precursors is an important process in the synthesis of nanomaterials and in the formation of aerosols in the atmosphere. Vapor-to-particle nucleation is a macroscopic process controlled by nanoscale particles (e.g., molecular clusters). Computational approaches to nucleation have been limited by the lack of a consistent theory of the process and by the lack of efficient approaches to simulate the properties of clusters relevant to nucleation. In this article, we focus on two advances that allow nucleation to be treated in a rigorous manner for molecular systems: dynamical nucleation theory permits a consistent treatment of the nucleation kinetics and aggregation-volume-bias Monte Carlo simulations provides an efficient approach to evaluate the thermodynamics of molecular clusters important in nucleation. The combination of these two approaches positions molecular computational approaches to make significant advances in our understanding of the mechanisms of nucleation, particularly in multiple component systems that play crucial roles in nanoscience applications and in the atmosphere.