Anal. Chem. 76, 2886-2892 (2004)
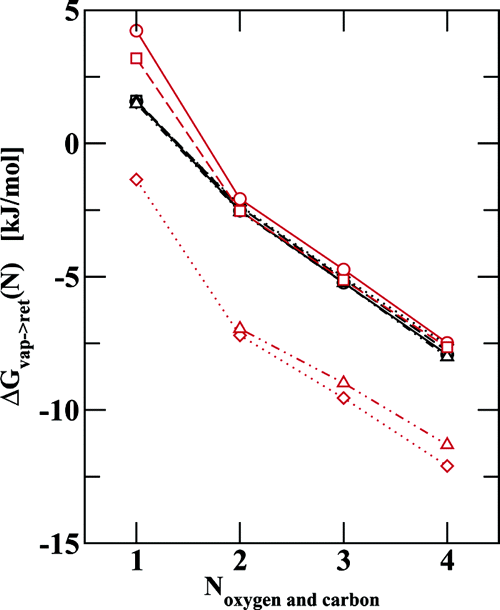
Various driving forces have been suggested to explain retention and selectivity in reversed-phase liquid chromatography (RPLC). To provide molecular-level information on the retention mechanism in RPLC, configurational-bias Monte Carlo simulations in the Gibbs ensemble were carried out for model systems consisting of three phases: an n-hexadecane retentive phase, a mobile phase with varying water−methanol composition, and a helium vapor phase as reference state. Liquid n-hexadecane functions as a model of a hydrophobic stationary phase, and a wealth of experimental data exists for this system. Gibbs free energies for solute transfers from gas to retentive phase, from gas to mobile phase, and from mobile to retentive phase were determined for a series of short linear alkanes and primary alcohols. Although the magnitude of the incremental Gibbs free energy of transfer for a methylene segment is always larger for the gas- to retentive-phase transfer than the gas- to mobile-phase transfer, it is found that the partitioning of alkanes and alkyl tail groups is mostly affected by the changes in the aqueous mobile phase that occur when methanol modifiers are added. In contrast, the partitioning of the alcohol headgroup is sensitive to changes in both the n-hexadecane and the mobile phases. In particular, it is found that hydrogen-bonded aggregates of methanol are present in the n-hexadecane phase for higher methanol concentrations in the mobile phase. These aggregates strongly increase alcohol partitioning into the retentive phase. The simulation data clearly demonstrate that due to modification of the retentive-phase hydrocarbons by solvent components, neither the solvophobic theory of RPLC, advocated by Horvath and co-workers, nor the lipophilic theory of RPLC, advocated by Carr and co-workers, can adequately describe the separation mechanism of the hexadecane model system of a retentive phase studied here nor the more complex situation present in actual RPLC systems.