J. Am. Chem. Soc. 127, 4722-4729 (2005)
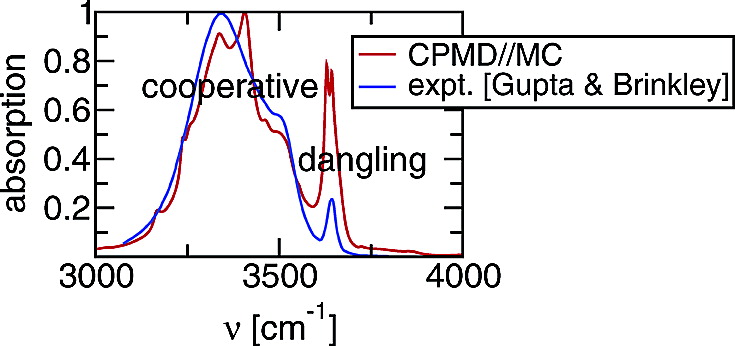
Fourier transform infrared spectroscopy is a popular method for the experimental investigation of hydrogen-bonded aggregates, but linking spectral information to microscopic information on aggregate size distribution and aggregate architecture is an arduous task. Static electronic structure calculations with an implicit solvent model, Car−Parrinello molecular dynamics (CPMD) using the Becke−Lee−Yang−Parr (BLYP) exchange and correlation energy functionals and classical molecular dynamics simulations for the all-atom version of the optimized parameters for liquid simulations (OPLS-AA) force field were carried out for an ensemble of 1-hexanol aggregates solvated in n-hexane. The initial configurations for these calculations were size-selected from a distribution of aggregates obtained from a large-scale Monte Carlo simulation. The vibrational spectra computed from the static electronic structure calculations for monomers and dimers and from the CPMD simulations for aggregates up to pentamers demonstrate the extent of the contribution of dangling or nondonating hydroxyl groups found in linear and branched aggregates to the “monomeric” peak. Furthermore, the computed spectra show that there is no simple relationship between peak shift and aggregate size nor architecture, but the effect of hydrogen-bond cooperativity is shown to differentiate polymer-like (cooperative) and dimer-like (noncooperative) hydrogen bonds in the vibrational spectrum. In contrast to the static electronic structure calculations and the CPMD simulations, the classical molecular dynamics simulations greatly underestimate the vibrational peak shift due to hydrogen bonding.