J. Phys. Chem. B 110, 3738-3746 (2006)
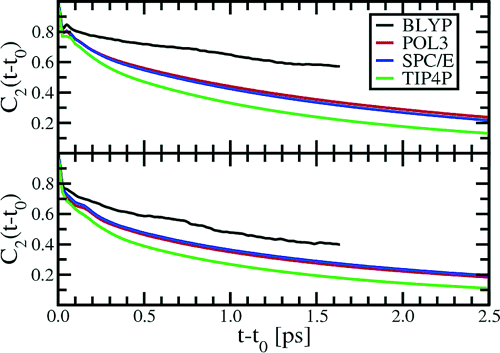
This research addresses a comprehensive particle-based simulation study of the structural, dynamic, and electronic properties of the liquid−vapor interface of water utilizing both ab initio (based on density functional theory) and empirical (fixed charge and polarizable) models. Numerous properties such as interfacial width, hydrogen bond populations, dipole moments, and correlation times will be characterized with identical schemes to draw useful conclusions on the strengths and weakness of the proposed models for interfacial water. Our findings indicate that all models considered in this study yield similar results for the radial distribution functions, hydrogen bond populations, and orientational relaxation times. Significant differences in the models appear when examining both the dipole moments and surface relaxation near the aqueous liquid−vapor interface. Here, the ab initio interaction potential predicts a significant decrease in the molecular dipole moment and expansion in the oxygen−oxygen distance as one approaches the interface in accordance with recent experiments. All classical polarizable interaction potentials show a less dramatic drop in the molecular dipole moment, and all empirical interaction potentials studied yield an oxygen−oxygen contraction as the interface is approached.