Cryst. Growth Des. 6, 1318-1323 (2006)
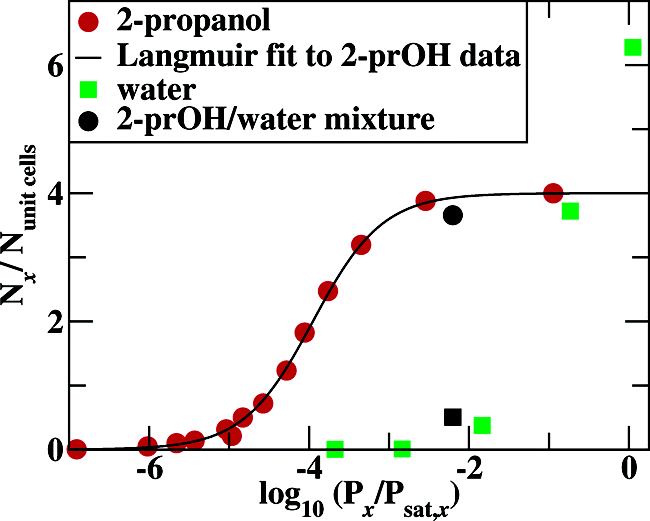
Monte Carlo simulations in the isotension−isothermal and Gibbs ensembles were carried out to investigate the structural properties of warfarin sodium 2-propanol and the ability of this compound to retain 2-propanol as a function of vapor pressure and in the presence of water. At full loading, the simulations yield a stable solvate structure that is in good agreement with the experimentally determined crystal structure. 2-Propanol is well-retained by the warfarin sodium host, and full loading is observed for partial pressures as low as 1% of its saturated vapor pressure, while water does not substantially replace 2-propanol from the crystalline interior even at 100% relative humidity. Radial distribution functions show strong binding of 2-propanol's hydroxyl hydrogen with the 2-keto oxygen and the oxygen atom of the coumarin ring of warfarin and strong binding of 2-propanol's oxygen to a single sodium ion. Thus, the spatial position of 2-propanol is highly confined, supporting the experimental observation that the 2:1 adduct of warfarin sodium and 2-propanol is a true solvate and not a clathrate. In contrast, water molecules would be less constrained and two water molecules would fit into 2-propanol's binding site, but there is no channel for the transport of water or 2-propanol molecules.