J. Chromatogr. A 1126, 373-380 (2006)
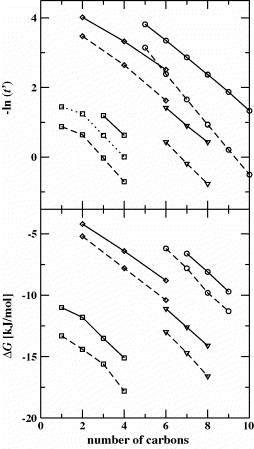
Configurational-bias Monte Carlo simulations in the isobaric–isothermal Gibbs ensemble were carried out to investigate the partitioning of normal alkanes, primary and secondary alcohols, symmetric alkyl ethers and arenes between a helium vapor phase and a polyethylene oxide stationary phase (MW=382 g mol−1). The united-atom version of the transferable potentials for phase equilibria force field was used to model all solutes, polyethylene oxide and helium. The Gibbs free energies of transfer and Kovats retention indices of the solutes were calculated directly from the partition constants at two different temperatures, 353 and 393 K. Chromatographic experiments on a Carbowax 20M retentive phase were performed for the same set of solutes and temperatures ranging from 333 to 413 K. The predicted retention indices for alcohols, ethers and arenes are overestimated by about 120, 70 and 20 retention index units, respectively, pointing to an overestimation of the first-order electrostatic interactions in the model system. Molecular-level analysis shows that hydrogen-bonding and dipole–dipole interactions lead to orientational ordering for the alcohol and ether analytes, whereas the weaker dipole–quadrupole interactions for the arene solutes are not sufficient to induce orientational ordering. The retention indices of alcohols and ethers decrease with increasing temperature because of the large entropic cost of hydrogen-bonding and orientational ordering. In contrast, the retention indices for arenes increase with increasing temperature because the entropic cost of cavity formation is smaller for arenes than for comparable alkanes.