J. Chem. Theor. Comp. 2, 1274-1281 (2006)
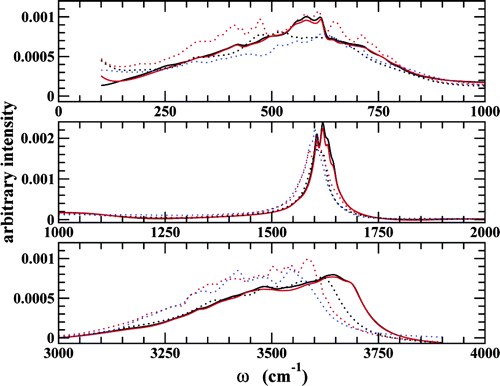
A series of 30 ps first principles molecular dynamics simulations in the microcanonical ensemble were carried out to investigate transport and vibrational properties of liquid water. To allow for sufficient sampling, the thermodynamic constraints were set to an elevated temperature of around 423 K and a density of 0.71 g cm-3 corresponding to the saturated liquid density for the Becke−Lee−Yang−Parr (BLYP) representation of water. Four simulations using the Car−Parrinello molecular dynamics (CPMD) technique with varying values of the fictitious electronic mass (μ) and two simulations using the Born−Oppenheimer molecular dynamics (BOMD) technique are analyzed to yield structural and dynamical information. At the selected state point, the simulations are found to exhibit nonglassy dynamics and yield consistent results for the liquid structure and the self-diffusion coefficient, although the statistical uncertainties in the latter quantity are quite large. Consequently, it can be said that the CPMD and BOMD methods produce equivalent results for these properties on the time scales reported here. However, it was found that the choice of μ affects the frequency spectrum of the intramolecular modes, shifting them slightly to regions of lower frequency. Using a value of μ = 400 au results in a significant drift in the electronic kinetic energy of the system over the course of 30 ps and a downward drift in the ionic temperature. Therefore, for long trajectories at elevated temperatures, lower values of this parameter are recommended for CPMD simulations of water.