Intl. J. Thermophys. 22, 111-122 (2001)
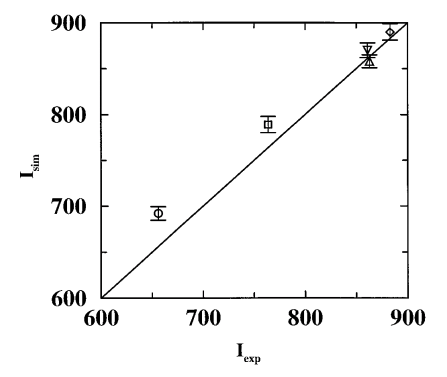
Accurate predictions of retention times, retention indices, and partition constants are a long sought-after goal for theoretical studies in chromatography. Although advances in computational chemistry have improved our understanding of molecular interactions, little attention has been focused on chromatography, let alone calculations of retention properties. Configurational-bias Monte Carlo simulations in the isobaric–isothermal Gibbs ensemble were used to investigate the partitioning of benzene, toluene, and the three xylene isomers between a squalane liquid phase and a helium vapor phase. The united-atom representation of the TraPPE (transferable potentials for phase equilibria) force field was used for all solutes and squalane. The Gibbs free energies of transfer and Kovats retention indices of the solutes were calculated directly from the partition constants (which were averaged over several independent simulations). While the calculated Kovats indices of benzene and toluene at T=403 K are significantly higher than their experimental counterparts, much better agreement is found for the xylene isomers at T=365 K.